Add nextflow pipeline
Nextflow Integration
Nextflow is a powerful and flexible workflow management system that supports various types of pipelines.
Overview
This brief document outlines the requirements and considerations for integrating Nextflow pipeline with Basepair. The integration will allow users to run their Nextflow pipelines on the Basepair platform, leveraging the benefits of cloud-based computing infrastructure and enhanced data management capabilities. Maximize your workflow efficiency with Basepair's versioning , allowing you to effortlessly switch between pipeline versions.
- Prerequisites
- Pipeline & Module YAML Creation
Custom Nextflow pipeline integration on basepair is almost ready to go live!
Direct FASTQ Upload to BP!
Upload your raw FASTQ files directly to BP via GUI/CLI/Basespace. No need to create a separate SampleSheet CSV file - we'll generate it automatically during analysis. Ready to test? Login to BP platform and kick off some analysis to validate your integration You'll be able to monitor the progress of your analysis and access the results when it's complete.
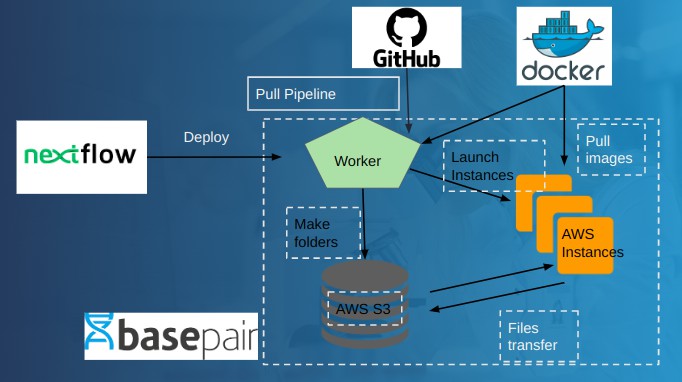
Prequisites
Get Started Quickly:
This document outlines the various methods to import your Nextflow pipeline on Basepair platform for efficient and streamlined analysis.
-
Package Your Pipeline
-
Public GitHub Repository: If your pipeline is publicly available on GitHub, Basepair can directly access and utilize it at the Module YAML level. This is the most straightforward approach for public pipelines.
-
Private GitHub Integration : Basepair has developed a feature that allows for direct integration with private GitHub repositories using a GitHub access token. This enables users to easily install the Basepair GitHub app through the "Integrations" tab within the "Organization Settings" section of their user profile on the Basepair platform
-
-
Database & Reference Files (Optional)
This section pertains to specific scenarios where your pipeline relies on:
- Proprietary databases
- Reference genome files If your pipeline necessitates these resources, you can upload them to the same S3 bucket you used for your pipeline archive. This ensures Basepair has all necessary components for successful analysis. Alternatively, you can leverage Basepair's pre-uploaded genome files. These files will be attached as metadata to each sample object, simplifying the process.
-
Docker Image Access Currently, Basepair requires access to your private Dockerfile stored in a secure location like Amazon ECR (Elastic Container Registry). This Dockerfile provides instructions for building the containerized environment necessary for your pipeline's execution. Basepair puts user-friendliness first, even for secure private images. New feature coming for streamlined, secure pulling docker images.
-
Basepair API Setup:
Refer to Basepair's documentation for comprehensive instructions on setting up API to interact with the platform
Pipeline & Module YAML Creation
Demo Module YAML , configure it based on your NF pipeline
=name: testname # Module name
type: testtype # Module type
version: v1.0.0
path: /home/ec2-user/bioinfo/infra/repository
command:
## Install Java and Nextflow
# This section install the required versions of java
and nextflow
- /home/ec2-user/bioinfo/infra/storage/sync_reflib.py
-k reflib/tool/install-java-on-worker.sh -d /home/ec2- user/tools/ -f
- sudo chmod +x /home/ec2-user/tools/install-java-on-
worker.sh && sudo sh /home/ec2-user/tools/install-java-
on-worker
- export NXF_VER=23.10.0 # Choose nextflow version to
use
- wget -qO- <https://get.nextflow.io> | bash
- sudo mv nextflow /usr/bin/
## Set Up AWS Variables
# This section configure the scripts to use your AWS credential
- REGION=$(jq '.repository.settings.region'
/home/ec2-user/.bpconfig.json | sed 's/"//g') ## For Private Git Repo
- python3.8 {{path}}/github_integration.py --url
https://github.com/gitaccount/reponame.git --clonedir
{{basedir}}/reponame/ --branch {{nfcode_version}} && cd
{{basedir}}/reponame/
# optional params --branch [release tag / branch
name]
## For Public Git Repo (Note:comment above private git repo)
# git clone -b {{nfcode_version}} --single-branch
git@github.com:gitaccount/reponame.git
{{basedir}}/reponame/ && cd {{basedir}}/reponame/
## SampleSheet Creation
# Below script will create a sample sheet csv input file , pass the column header name in below arguments as
your nf scripts expected :-
# 1. --samplename { colname for samples to prepare samplesheet }
# 2. --forcolname { colname for forward fastq to
prepare samplesheet }
# 3. --revcolname { colname for reverse fastq to
prepare samplesheet}
# 4. --extra_columnn value --extra_datan value { 38
colname for extra column and data value where can be
integer }
41 - |
/home/ec2-user/bioinfo/bioinfo/nf_samplesheet.py -
-sampleids {{all_sample_ids}} --samplename 'sample' --
forcolname '' --revcolname '' --extra_column1 '' --
extra_data1 '' --outdir {{basedir}}/
## Run Nextflow CMD
# Add required params to nextflow run command with
default values as well as add them to Inputs section so
user can change values at the analysis page.
50 - |
nextflow run main.nf -profile docker --input
{{basedir}}/samplesheet.csv --outdir
{{basedir}}/results/ --max_cpus {{max_cpus}}
# NOTE:- To run on AWS batch add --profile
awsbatch,docker --awsregion $REGION --awsqueue job-queue-
prod
# This section includes the inputs/params (i.e
str,int,float,option type) user can provide to command
section
inputs:
nfcode_version:
62 val: v1.0.1
type: option
show: true # make it false if don't want to show this
at analysis page
label: nf code pipeline version
options:
68 - v1.0.0
69 - v1.0.1
70 - v1.0.2
71 - v1.0.3
help: "select version to run nf pipeline"
max_cpus:
val: 24
type: int
min: 8
max: 36 label: Max CPU
show: false
## This section includes the outputs (i.e file,zip,folder) options to save the results and display them at analysis page
# Add or remove outputs filed based on pipeline requirements.
outputs:
input_csv: type: file val: ''
action: template
formula: _{{basedir}}/samplesheet.csv summary_html:
type: file
val: ''
action: template formula:
_{{basedir}}/results/summary_report_final.html
tags: [report, html] zip_outdir:
type: file
val: 'Output.zip' action: template dir_action: template
dir_formula: _{{basedir}}/results/ dir_val: _{{basedir}}/results/
formula: _{{basedir}}/Output.zip
Push Module YAML to basepair database
# Module create command creates a new module in the BPPython
database and returns id which needs to be added to the
top of module YAML eg. id: 12345.
basepair module create --file
~/p athtomodule/modulename.yaml
5 # Module update command if changes done at module level needs to update on BP database.
basepair module update --file
~/pathtomodule/modulename.yaml
Demo Pipeline YAML, configure it based on your NF pipeline requirements
name: 'pipelinename' #name of pipeline summary: |
'Summary about pipeline'
description: |
'Description about pipeline' datatype: dna-seq # {atac-seq,chip-
seq,crispr,cutnrun,cutntag,dna-seq,other,panel,rna-
seq,scrna-seq,small-rna-seq,snap-chip,wes,wgs} visibility: private # choose between public/private
# Choose instance from below list based on MAX Memory and CPU defined in nextflow config for docker profile.
# Instane types: "c1.medium","c3.2xlarge","c3.4xlarge","c3.8xlarge","c3.la rge","c3.xlarge","c4.8xlarge","c5d.18xlarge","c5d.24xlarg e","c5d.2xlarge","c5d.4xlarge","c5d.9xlarge","c5d.large",
"c5d.xlarge","c6gd.large","i3.16xlarge","i3.2xlarge","i3. 4xlarge","i3.8xlarge","i3en.xlarge","m1.large","m1.medium ","m1.small","m1.xlarge","m2.2xlarge","m2.4xlarge","m3.2x
large","m3.large","m3.medium","m3.xlarge","m5d.12xlarge", "m5d.2xlarge","m5d.4xlarge","m5d.8xlarge","m5d.large","m5 d.xlarge","m6gd.medium","r3.2xlarge","r3.4xlarge","r3.lar
ge","r3.xlarge","t3.micro","t3.nano","t4g.nano","x1e.16xl
arge"
infra:
instance_type: c1.medium
save_app_node_id: 'save' tags: [nf, pipelinename]
21
validation:
required:
filetypes:
- fastq
genome: false # True if basepair genome files will be
going to use
num_samples: '1' # Default '1' analysis work for
single sample at a time where '1+' indicates more than 1
sample.
num_controls: '0' # Add control sample counts
paired: true # switch to false for single end data
datatype:
- dna-seq
35
edges:
- parent_node_id: '-1'
app_node_id: 'start'
39
- parent_node_id: 'start'
app_node_id: 'modulename'
42
- parent_node_id: 'modulename'
app_node_id: 'stop'
45
nodes:
'save':
app_id: '9'
info:
bucket: bucket
51
'start':
app_id: '5'
info:
dirname: compute_basedir
56
'modulename':
app_id: 'Id used at module creation'
info:
num_threads: num_threads #fetched from analysis api
memory: memory #fetched from analysis api
bucket: bucket #fetched from analysis api
all_sample_ids: all_sample_ids #fetched from
analysis api
storage_basedir: storage_basedir #fetched from analysis api
basedir: compute_basedir #fetched from analysis api genome_name: genome_id #fetched from analysis api
fasta: genome_id #fetched from analysis api genome_id: genome_id #fetched from analysis api slug: slug #fetched from analysis api
'stop': app_id: '22' info:
compute_basedir: compute_basedir
Push Pipeline YAML to basepair database
# Pipeline create command: creates a new pipeline in thPyethon BP database and returns id which needs to be added to the top of pipeline YAML eg. id: 10000
basepair pipeline create --file
~/pathtopipeline/pipelinename.yaml
5
# Pipeline update command:
basepair pipeline update --file
~/pathtopipeline/pipelinename.yaml -u 10000